At WiseGEEK, we're committed to delivering accurate, trustworthy information. Our expert-authored content is rigorously fact-checked and sourced from credible authorities. Discover how we uphold the highest standards in providing you with reliable knowledge.
What are Lipidoses?
The general term lipidoses, describes any one of more than 30 inherited metabolic disorders causing abnormal accumulations of fats, oils or steroids in tissues, resulting from the body‘s inability to properly break down these substances. Certain patients exhibit symptoms of lipid storage disorders shortly after birth, while other disorders may not produce symptoms until individuals reach adulthood. Symptoms also vary depending on what part of the body is affected by the disorder.
Under normal circumstances, at birth individuals produce the appropriate enzymes required to metabolize and utilize various food substances. Persons born with metabolic disorders typically lack the genetic coding responsible for making sufficient amounts of lipid metabolizing enzymes, or the manufactured enzymes malfunction. Parents carrying a defective gene are generally without symptoms of the disorder. Health care providers consider lipidoses relatively uncommon, but some of these inherited disorders appear more frequently.

Variations of Gaucher disease, for example, affect females or males in many parts of the world, and specialists consider the disorder one of the more common forms of lipidoses. Both parents must carry the gene before passing the malady onto offspring. Symptoms occur when abnormal fat accumulates and displaces healthy cells in the bones, liver, spleen and nervous system. Patients generally experience a wide array of symptoms, which include abdominal swelling from liver and spleen involvement, stunted growth or fractures from affected bone tissue, and fatigue from anemia due to the tendency to bruise or bleed easily. Treatment involves symptomatic relief and enzyme replacement.

Research suggests that one in 250 people carry the gene for Tay-Sachs disease, though the disorder more prominently affects persons of Jewish descent. Abnormal amounts of fatty acids accumulate in the nervous systems of children born with this form of lipidoses. Children afflicted with Tay-Sachs become blind, lose physical coordination and have mental developmental hindrances. As the disease progresses, the disorder causes seizures, difficulty breathing and an inability to swallow, ultimately culminating in death. Treatment typically involves alleviation of symptoms, though research continues.

Cerebrotendinous xanthomatosis is a lipidoses characterized by abnormal accumulations of cholesterol throughout the body. The disorder commonly manifests with chronic diarrhea during infancy; cataracts during childhood; and the presence of xanthomatosis, or cholesterol deposits, under the skin and various other locations during young adulthood. Patients often experience delayed mental development accompanied by various psychiatric symptoms, along with muscular spasms, tremors and seizure activity. Treatment involves symptomatic relief and enzyme supplementation, which aids in metabolizing cholesterol.
AS FEATURED ON:
AS FEATURED ON:

-
![A child with Tay-Sachs will typically appear healthy and normal for the first few months after birth.]() By: Patricia MarksA child with Tay-Sachs will typically appear healthy and normal for the first few months after birth.
By: Patricia MarksA child with Tay-Sachs will typically appear healthy and normal for the first few months after birth. -
![Patients with Tay-Sachs who are unable to swallow food may have to use a feeding tube.]() By: sudok1Patients with Tay-Sachs who are unable to swallow food may have to use a feeding tube.
By: sudok1Patients with Tay-Sachs who are unable to swallow food may have to use a feeding tube. -
![Metabolic disorders are often inherited.]() By: Monkey BusinessMetabolic disorders are often inherited.
By: Monkey BusinessMetabolic disorders are often inherited. -
![As Tay-Sachs disease progresses, the disorder causes difficulty breathing and an inability to swallow.]() By: thepooAs Tay-Sachs disease progresses, the disorder causes difficulty breathing and an inability to swallow.
By: thepooAs Tay-Sachs disease progresses, the disorder causes difficulty breathing and an inability to swallow. -
![Lipidoses patients experience a wide array of symptoms, including abdominal swelling from liver and spleen involvement.]() By: stockshoppeLipidoses patients experience a wide array of symptoms, including abdominal swelling from liver and spleen involvement.
By: stockshoppeLipidoses patients experience a wide array of symptoms, including abdominal swelling from liver and spleen involvement. -
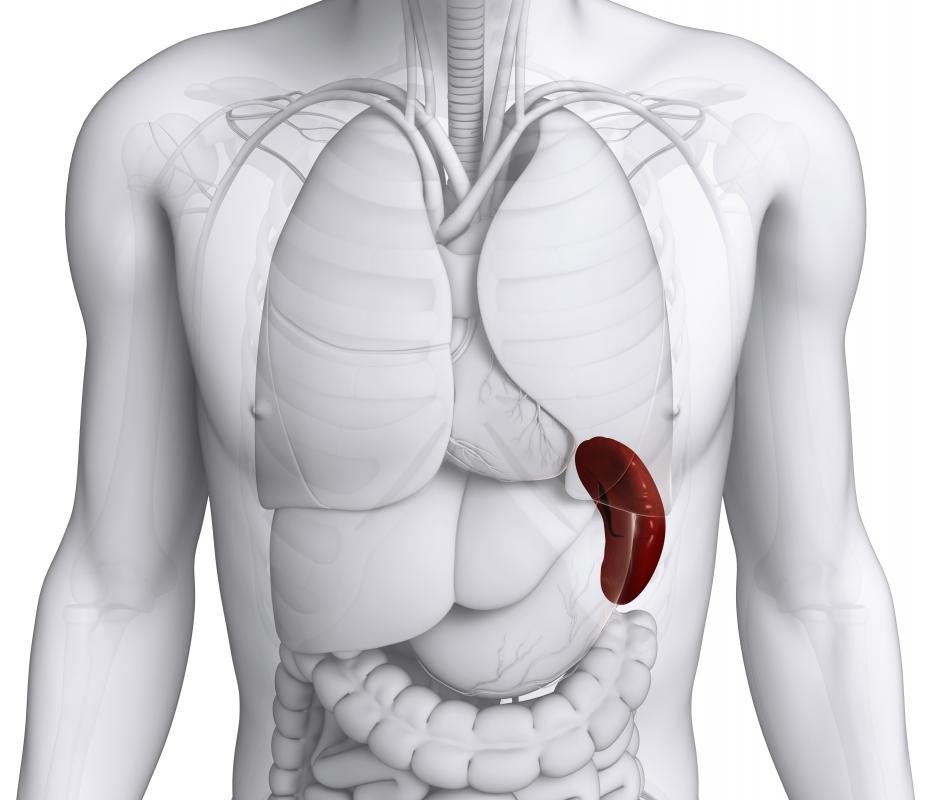
![Gaucher disease may cause swelling due to spleen enlargement.]() By: pixdesign123Gaucher disease may cause swelling due to spleen enlargement.
By: pixdesign123Gaucher disease may cause swelling due to spleen enlargement.





Discuss this Article
Post your comments