At WiseGEEK, we're committed to delivering accurate, trustworthy information. Our expert-authored content is rigorously fact-checked and sourced from credible authorities. Discover how we uphold the highest standards in providing you with reliable knowledge.
What is Wolman Disease?
Wolman disease is one of a family of disorders called lipid storage diseases. This type of disorder is caused by a genetically-inherited defect in one or more enzymes involved in lipid metabolism. Most of these disorders are autosomal recessive, which means both parents have a copy of the defective gene, and an affected child inherits the defective gene from each parent. Wolman disease is also called Wolman’s disease or Wolman’s syndrome and may also be known as acid lipase deficiency. Most lipid storage diseases are very rare in all populations; in the case of Wolman disease, the frequency is estimated to be 1 in 350,000 births.
Wolman’s disease is caused by a mutation in a gene called lysosomal acid, lipase A, or LIPA. This gene codes for a protein called lysosomal acid lipase. This protein is an enzyme which plays an instrumental role in breaking down lipids for use or storage. When the gene is defective, the body cannot metabolize certain types of fats. As a result, triglycerides and cholesterol build up within the body and are deposited in the adrenal glands, spleen, liver, intestines, and lymph nodes.

Lysosomal acid lipase deficiency causes symptoms related to the effects of fatty deposits in organs, and to the fact that inability to metabolize fats causes severe malnutrition. Symptoms of Wolman disease may appear as early as one week after birth. Possible symptoms include enlarged spleen, enlarged liver, jaundice, vomiting, diarrhea, anemia, little or no weight gain, and poor muscle tone.

Only a small number of lipid storage disorders are treatable. Wolman's disease is not one of these, and is considered a fatal disease of infancy, as most children die during their first year of life. Currently there is no cure, and no treatments which can reverse the effects of the gene mutation. Treatments for this disease focus on managing symptoms such as anemia and malnutrition.

One of the most common Wolman disease treatments is the use of intravenous nutritional supplements that bypass the digestive system. Another common therapy is blood transfusions for the treatment of anemia. There are no treatments which can prevent the build-up of lipids in organs, however; because of this, some infants with the disease eventually require surgery to remove an enlarged spleen. If fat deposition reduces adrenal gland function, medications can be given to replace the hormones the glands would normally secrete.

There is one recorded instance of a treatment for Wolman’s disease actually providing a long term benefit for the person with the condition. In this case the treatment was a bone marrow transplant, and caused the disease to enter a remission state in the person who received the treatment. This single success means there is potential for bone marrow transplant to become a standard treatment for the disease, but as yet the successful result has not been replicated in a second patient.
AS FEATURED ON:
AS FEATURED ON:

-
![An enlarged spleen may be a symptom of Wolman disease.]() By: CLIPAREA.comAn enlarged spleen may be a symptom of Wolman disease.
By: CLIPAREA.comAn enlarged spleen may be a symptom of Wolman disease. -
![Anemia may be a symptom of Wolman disease.]() By: joshyaAnemia may be a symptom of Wolman disease.
By: joshyaAnemia may be a symptom of Wolman disease. -
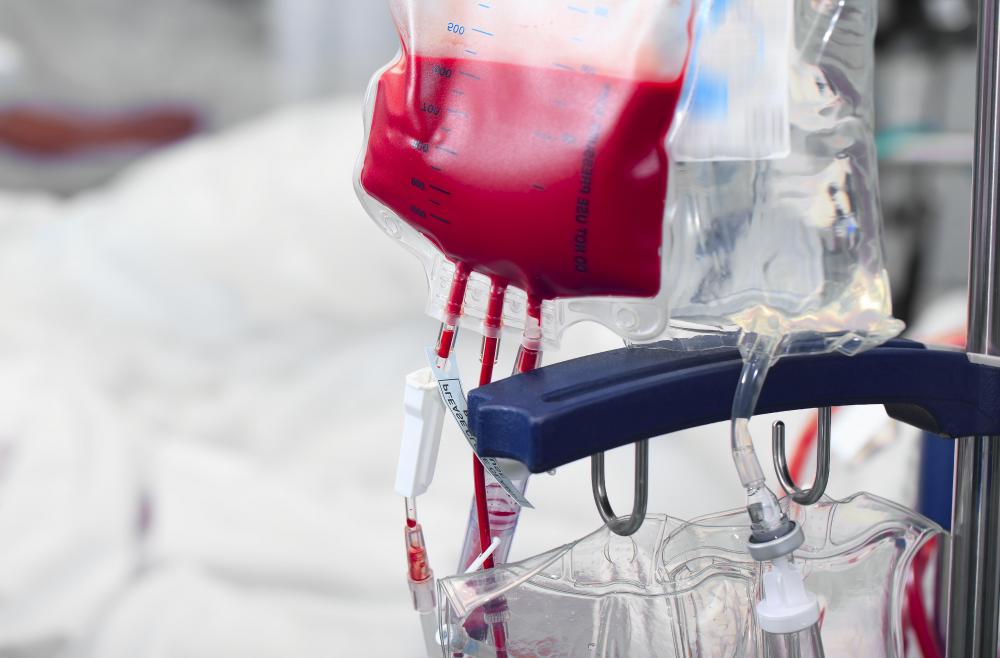
![Blood transfusions are a common treatment of Wolman disease.]() By: sudok1Blood transfusions are a common treatment of Wolman disease.
By: sudok1Blood transfusions are a common treatment of Wolman disease. -
![An enlarged liver may be a symptom of Wolman disease.]() By: maya2008An enlarged liver may be a symptom of Wolman disease.
By: maya2008An enlarged liver may be a symptom of Wolman disease. -
![Triglycerides and cholesterol build up within the body and are deposited in the adrenal glands as a result of Wolman disease.]() By: maya2008Triglycerides and cholesterol build up within the body and are deposited in the adrenal glands as a result of Wolman disease.
By: maya2008Triglycerides and cholesterol build up within the body and are deposited in the adrenal glands as a result of Wolman disease.




Discussion Comments
Does anyone on this website know how a day would be like for someone with this disorder? What they eat, time medications are taken, or at least how the family is affected; it would help me out a lot, thanks!
Post your comments