At TheHealthBoard, we're committed to delivering accurate, trustworthy information. Our expert-authored content is rigorously fact-checked and sourced from credible authorities. Discover how we uphold the highest standards in providing you with reliable knowledge.
What is Interstitial Pulmonary Fibrosis?
Interstitial pulmonary fibrosis is a form of interstitial lung disease characterized by the thickening and scarring of lung tissue. Frequently diagnosed as an idiopathic condition, meaning there is no known or obvious cause for its development, interstitial pulmonary fibrosis is a noninfectious, chronic disorder. Treatment for this form of pulmonary fibrosis generally involves the administration of steroidal medications to manage symptoms.
The lungs contain numerous airways, known as bronchioles, that culminate in the formation of air sacs called alveoli. Within these air sacs are capillaries that work to add oxygen to blood and remove carbon dioxide. In the presence of pulmonary fibrosis, the alveoli are irreversibly damaged due to the formation of scar tissue that essentially paralyzes alveoli function. As a result, the body is unable to repair the damage and breathing becomes more difficult.

There is no known, definitive cause for the development of interstitial pulmonary fibrosis. It has been asserted that the inhalation of certain toxins and pollutants, such as asbestosis or silicosis, may contribute to the onset of this debilitating condition. Individuals with a history of radiation exposure, tuberculosis, or autoimmune disease may also be at an increased risk for becoming symptomatic. Those who have been diagnosed with gastroesophageal reflux disease (GERD) are considered to possess an increased risk for pulmonary fibrosis.

The most pronounced symptom during the early stages of disease development is the impaired oxygenation of the blood within the lungs. Oxygen deprivation can induce fatigue, shortness of breath, and a general feeling of uneasiness. Additional symptoms associated with interstitial pulmonary fibrosis include joint discomfort, unintentional weight loss, and persistent cough. Breathing difficulty that manifests with disease onset gradually progresses with time. Symptom onset may be acute or gradually progress over time.

There are several diagnostic tests that may be administered to confirm a diagnosis of pulmonary fibrosis. In order to make a proper diagnosis, other medical conditions such as asthma and chronic obstructive pulmonary disease (COPD) must be ruled out. Once a complete medical history is taken and a physical examination is conducted, an individual may be referred for a battery of additional tests.

Imaging testing, such as a high-resolution computerized tomography (HRCT) scan and chest X-ray, may be performed to evaluate the condition of the lungs and determine the extent of scarring that may be present. Pulmonary function tests may be administered to assess the functionality of an individual’s lungs, including lung volume. A sample of lung tissue, known as a biopsy, may also be taken to further support a diagnosis of pulmonary fibrosis.

There is no cure for interstitial pulmonary fibrosis; therefore, treatment is centered on alleviating symptoms and slowing the progression of the disease. Individuals may be prescribed corticosteroids, such as prednisone, to ease inflammation and improve lung function. Additional immunosuppressive medications may be utilized to supplement the use of corticosteroids, though their use may result in serious side effects, including glaucoma and impaired red blood cell production.

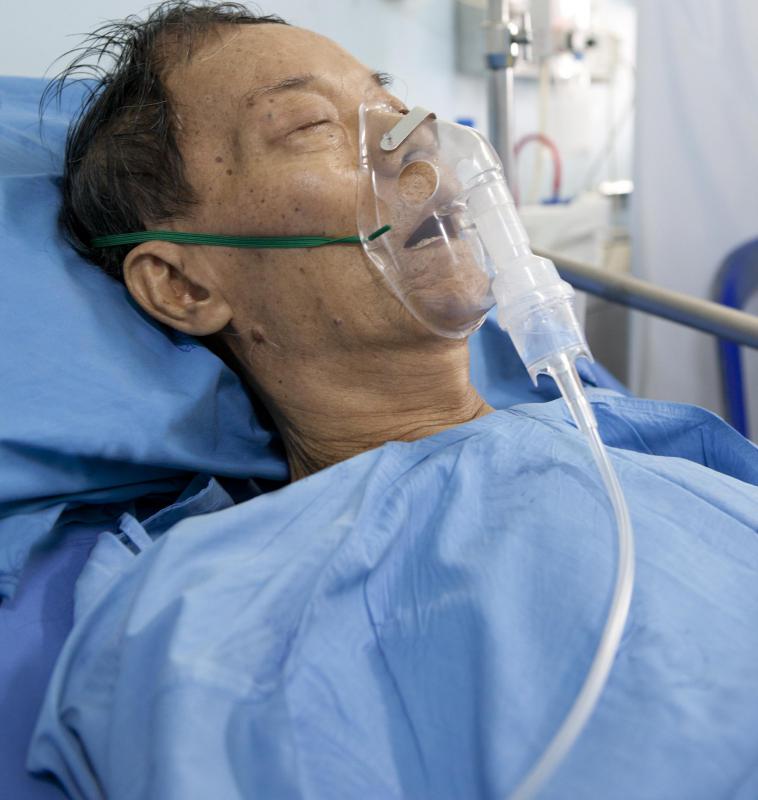
Oxygen therapy may be utilized to alleviate breathing difficulty and ease complications, such as lightheadedness and dizziness resulting from oxygen deprivation. Individuals in the advanced stages of pulmonary fibrosis who have not responded well to traditional treatments may undergo lung transplantation. Strict qualifications must be met to be considered for lung transplantation, including demonstrating that one is willing to follow through with post-operative treatments and rehabilitation requirements, and demonstrate the patience and understanding necessary while awaiting a donor.
Complications associated with interstitial pulmonary fibrosis include hypoxemia, or low blood-oxygen levels, and pulmonary hypertension. Individuals with this condition are also at an increased risk for developing heart and respiratory failure. Risk factors associated with the onset of pulmonary fibrosis include smoking, exposure to environmental and occupational hazards, and advanced age. Idiopathic interstitial pulmonary fibrosis may be triggered by smoking and exposure to certain viral infections. Individuals with a familial history of pulmonary fibrosis may also be at a higher risk for developing symptoms.
AS FEATURED ON:
AS FEATURED ON:

-
![Pulmonary circulation is the movement of blood from the heart, to the lungs, and back to the heart again.]() By: silotoPulmonary circulation is the movement of blood from the heart, to the lungs, and back to the heart again.
By: silotoPulmonary circulation is the movement of blood from the heart, to the lungs, and back to the heart again. -
![Chest x-rays may be needed to evaluate the condition of the lungs.]() By: PetrikChest x-rays may be needed to evaluate the condition of the lungs.
By: PetrikChest x-rays may be needed to evaluate the condition of the lungs. -
![Individuals might be prescribed corticosteroids, such as prednisone, to ease inflammation and improve lung function.]() By: Thirteen Of ClubsIndividuals might be prescribed corticosteroids, such as prednisone, to ease inflammation and improve lung function.
By: Thirteen Of ClubsIndividuals might be prescribed corticosteroids, such as prednisone, to ease inflammation and improve lung function. -
![Patients with interstitial pulmonary fibrosis are at an increased risk for heart and respiratory failure.]() By: khunaspixPatients with interstitial pulmonary fibrosis are at an increased risk for heart and respiratory failure.
By: khunaspixPatients with interstitial pulmonary fibrosis are at an increased risk for heart and respiratory failure. -
![Those with GERD may be at risk for developing interstitial pulmonary fibrosis.]() By: bilderzwergThose with GERD may be at risk for developing interstitial pulmonary fibrosis.
By: bilderzwergThose with GERD may be at risk for developing interstitial pulmonary fibrosis. -
![Interstitial pulmonary fibrosis is characterized by the thickening and scarring of lung tissue.]() By: Aleksandar TodorovicInterstitial pulmonary fibrosis is characterized by the thickening and scarring of lung tissue.
By: Aleksandar TodorovicInterstitial pulmonary fibrosis is characterized by the thickening and scarring of lung tissue.





Discuss this Article
Post your comments