At TheHealthBoard, we're committed to delivering accurate, trustworthy information. Our expert-authored content is rigorously fact-checked and sourced from credible authorities. Discover how we uphold the highest standards in providing you with reliable knowledge.
What is a Coagulation Cascade?
A coagulation cascade is the process by which the body forms blood clots to prevent excess blood loss. Coagulation begins with the extrinsic pathway, which activates clotting as a result of tissue injury, or the intrinsic pathway, which forms clots in response to abnormalities in the wall of a blood vessel in the absence of tissue injury. The extrinsic and intrinsic pathways start differently and then both progress to the combined pathway. Both pathways depend on multiple clotting factors, which are represented by the Roman numerals I through XIII. These clotting factors are present in the body in inactive forms until they are activated during the coagulation cascade.
The extrinsic pathway is the most frequent and most familiar of the two pathways of the coagulation cascade. When a tissue injury such as a cut or a contusion causes bleeding, the injured blood vessels constrict. Then a glycoprotein called tissue factor, or factor III, is released from inside the damaged tissue. Next, a protein in the blood called factor VII binds to the tissue factor, and this bond activates the factor VII, converting it to factor VIIa. Factor VIIa then cleaves factor X molecules circulating in the blood to form factor Xa. From this point on, the extrinsic and intrinsic pathways are the same and are known as the combined pathway.

The intrinsic pathway of the coagulation cascade begins when blood comes into contact with the collagen in the damaged wall of a blood vessel. This causes factor XII, the Hageman factor, to convert into its active form, factor XIIa. Factor XIIa converts factor XI into factor XIa, which converts factor IX into factor IXa. Hemophilia B is a genetic bleeding disorder characterized by a deficiency in factor IX. Factor IXa converts factor X into factor Xa; this is the point at which the intrinsic pathway joins the combined pathway.

In the combined pathway, factor Xa activates factor II, or prothrombin, to form factor IIa, or thrombin. Thrombin activates the blood's platelets and causes them to gather together at the affected site and form a platelet plug. A substance called von Willebrand factor (vWF) makes it possible for platelets to stick to the injured site. A deficiency of vWF causes von Willebrand disease, the most common inherited bleeding disorder.

Thrombin cleaves fibrinogen molecules, or factor I molecules, to form a fibrous protein called fibrin, or factor Ia. The fibrin forms a net that catches the platelet plug and forms a clot. Thrombin also continuously converts factors V and VIII to their activated forms, which keeps the coagulation cascade going and accelerates the process. Hemophilia A, the most common type of hemophilia, is a genetic bleeding disease characterized by a deficiency in factor VIII.

A key component of the coagulation cascade is to dissolve the clot after it is no longer needed. After injury, blood vessel walls release inactive tissue plasminogen activator (tPA). The tPA comes in contact with the fibrin in the clot and becomes active. The active tPA activates a substance called plasminogen, which is already present within the clot. The plasminogen turns into plasmin, which dissolves the clot.

If a person bleeds excessively, doctors can run blood tests that check for the coagulation factors and detect bleeding disorders such as von Willebrand disease or hemophilia. Conditions such as liver disease and leukemia also can interfere with the coagulation cascade and cause excessive bleeding or bruising, so a person who develops these symptoms should see a doctor. An infection that reaches the blood can cause a condition known as sepsis, which leads to uncontrolled activation of the coagulation cascade all throughout the body and is the most frequent cause of death in non-cardiac intensive care patients. The coagulation cascade is a complicated system, and each person's survival depends on its proper function.
AS FEATURED ON:
AS FEATURED ON:

-
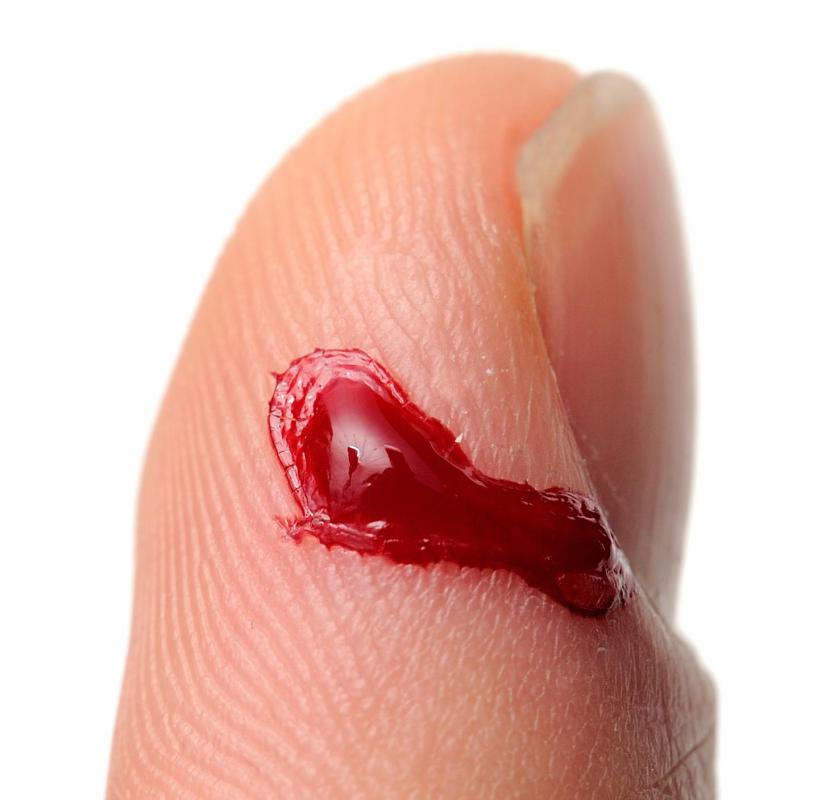
![Blood clots are the result of a coagulation cascade.]() By: clearviewstockBlood clots are the result of a coagulation cascade.
By: clearviewstockBlood clots are the result of a coagulation cascade. -
![Coagulation cascade is the process by which the body forms blood clots.]() By: VidadyCoagulation cascade is the process by which the body forms blood clots.
By: VidadyCoagulation cascade is the process by which the body forms blood clots. -
![Individuals with hemophilia are advised against donating blood.]() By: Gina SandersIndividuals with hemophilia are advised against donating blood.
By: Gina SandersIndividuals with hemophilia are advised against donating blood. -
![Hypnotherapy may be used to treat hemophilia.]() By: James SteidlHypnotherapy may be used to treat hemophilia.
By: James SteidlHypnotherapy may be used to treat hemophilia. -
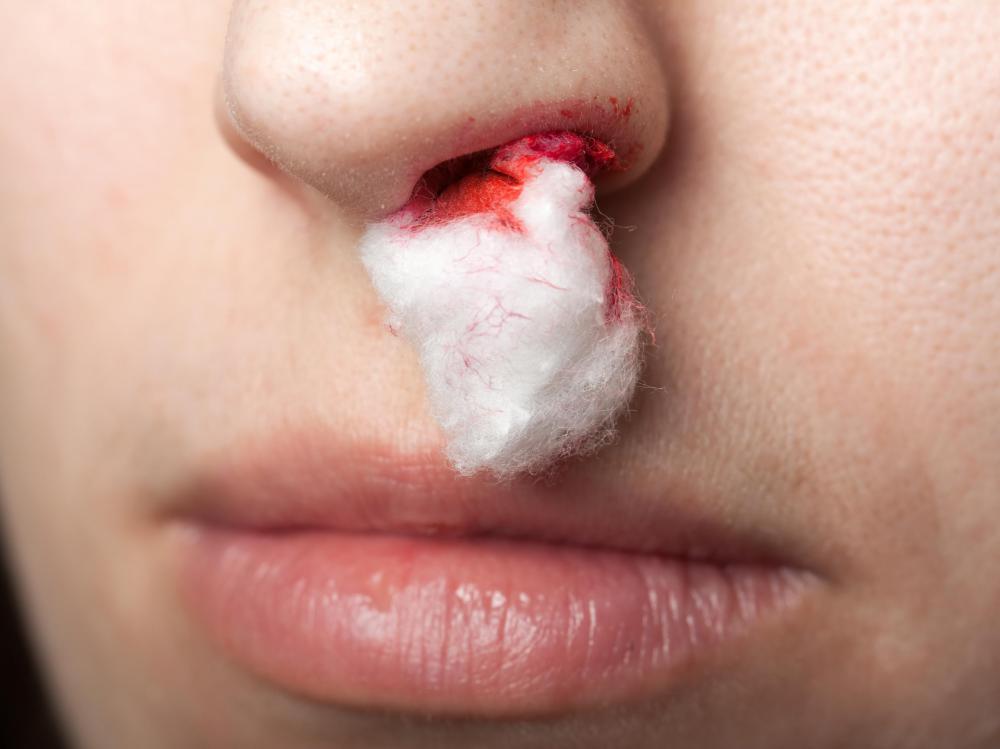
![Problems with the blood clotting process may cause nosebleeds.]() By: ia_64Problems with the blood clotting process may cause nosebleeds.
By: ia_64Problems with the blood clotting process may cause nosebleeds. -
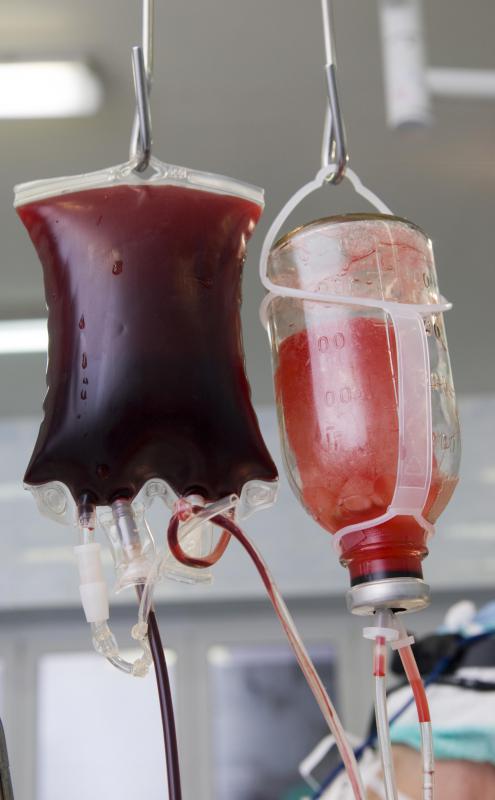
![A person who bleeds excessively may require a blood transfusion.]() By: Max TacticA person who bleeds excessively may require a blood transfusion.
By: Max TacticA person who bleeds excessively may require a blood transfusion.



Discuss this Article
Post your comments