At WiseGEEK, we're committed to delivering accurate, trustworthy information. Our expert-authored content is rigorously fact-checked and sourced from credible authorities. Discover how we uphold the highest standards in providing you with reliable knowledge.
What are the Different Types of PKD?
Polycystic kidney disease, commonly referred to as PKD or PCKD, is a genetic condition in which the kidneys develop multiple cysts. A cyst is an abnormal sac in which gas, liquid, or semi-solid material is enclosed. The cysts that occur as a result of PKD are filled with fluid, vary in size, and cause a dramatic enlargement of the kidneys. Though it primarily affects the kidneys, PKD can also create cysts in the liver, colon, and pancreas and damage the blood vessels, heart, and brain. There are two types of PKD, autosomal dominant and autosomal recessive.
The kidney is a paired organ located in the posterior, or toward the back, of the abdomen. These organs are responsible for producing urine, filtering the blood, reabsorption of sugars and salt into the bloodstream, and managing the body’s water concentration, amongst other functions. The kidneys of patients with PKD progressively lose their ability to perform these functions, sometimes culminating in kidney failure. Usually, this takes many years to occur.

People who have PKD often do not notice any symptoms. Symptoms that may develop include high blood pressure, headaches, blood in urine, kidney stones, back or side pain, abdominal swelling, frequent urination, infections in the urinary tract or kidneys, and, at end stages, kidney failure. In the event of kidney failure, patients may require a kidney transplant or dialysis, an artificial replacement for kidney functions.

The two types of PKD, autosomal dominant and autosomal recessive, are divided based on the different genetic defects that cause PKD. Autosomal dominant polycystic kidney disease (ADPKD) is by far the more common of the two. This defect is passed via a dominant gene, so that if one parent has the disease, the child has a fifty percent chance of developing the disorder as well. ADPKD was called adult PKD in the past, because it often presents symptoms between the ages of thirty and forty. Children, however, may also develop autosomal dominant PKD.

The defect that causes autosomal recessive polycystic kidney disease (ARPKD) is passed by a recessive gene. Because the gene is recessive, if one parent carries the defect and the other parent does not, the defective gene will be overridden by the healthy gene. If both parents carry the abnormal recessive gene, however, the child has a twenty-five percent chance of developing ARPKD. Unlike ADPKD, which may stem from one of two possible defective genes, only one gene is associated with ARPKD. Symptoms of ARPKD usually present in infancy, but may not develop until the late childhood or early teenage years.

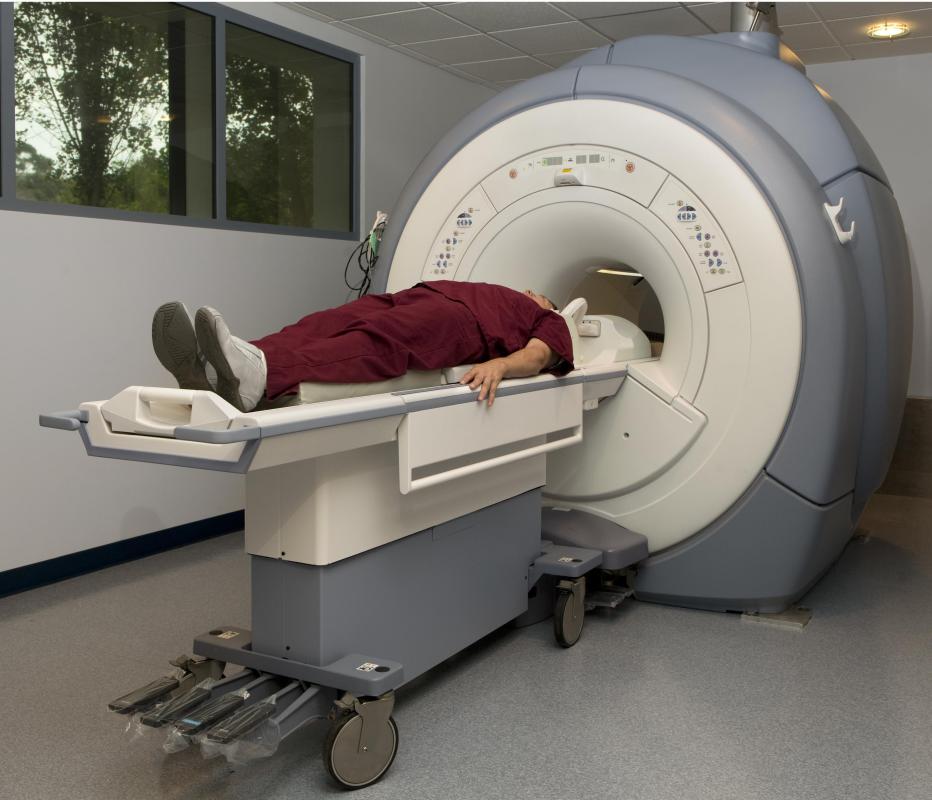
A physician may unintentionally discover PKD or purposefully search for PKD with imaging technology. Typically, these exams include an ultrasound scan, a computerized tomography (CT) scan, or a magnetic resonance imaging (MRI) scan. In some instances, particularly when a family member is donating a kidney, genetic testing may also be included in the diagnosis phase. Treatment for the symptoms and complications of the PKD will then be determined based on the size, number, and locations of the cysts and the symptoms the patient is experiencing. Treatment may include drugs to control blood pressure, surgery to drain cysts, antibiotic treatment of infections, pain medications, and continued monitoring of the affected areas.
AS FEATURED ON:
AS FEATURED ON:

-
![PKD is associated with kidney stones.]() By: erik gouldPKD is associated with kidney stones.
By: erik gouldPKD is associated with kidney stones. -
![An example of a healthy kidney and one with polycystic kidney disease.]() By: AlilaAn example of a healthy kidney and one with polycystic kidney disease.
By: AlilaAn example of a healthy kidney and one with polycystic kidney disease. -
![Side pain may be a sign of PKD.]() By: JackFSide pain may be a sign of PKD.
By: JackFSide pain may be a sign of PKD. -
![An MRI scan may be used to diagnose PKD.]() By: James SteidlAn MRI scan may be used to diagnose PKD.
By: James SteidlAn MRI scan may be used to diagnose PKD. -
![A renal ultrasound may be conducted to confirm the presence of PKD.]() By: Klaus EppeleA renal ultrasound may be conducted to confirm the presence of PKD.
By: Klaus EppeleA renal ultrasound may be conducted to confirm the presence of PKD. -
![If PKD leads to kidney failure, an advanced treatment like dialysis becomes necessary.]() By: Tyler OlsonIf PKD leads to kidney failure, an advanced treatment like dialysis becomes necessary.
By: Tyler OlsonIf PKD leads to kidney failure, an advanced treatment like dialysis becomes necessary.





Discuss this Article
Post your comments